Changer l'histoire : Comment nitisinone redonne de l'espoir aux patients HT-1 et AKU
Dernière mise à jour : 15 janvier 2024


Vous pouvez légalement accéder à de nouveaux médicaments, même s'ils ne sont pas approuvés dans votre pays.
Apprendre commentLorsqu'on est confronté à une maladie rare, il peut être aussi important de trouver des encouragements que de trouver un traitement. Nous sommes là pour vous aider dans les deux cas, en commençant par l'exemple de la tyrosinémie héréditaire de type 1 (HT-1) et de l'alkaptonurie (AKU).
Même si l'HT-1 et l'AKU touchent environ 1 personne sur 100 000, des traitements tels que nitisinone ont contribué à modifier le pronostic pour les patients. Grâce aux programmes d'accès précoce (en particulier en Inde, au Pakistan, au Bangladesh et au Soudan), davantage de patients peuvent accéder au traitement, quelle que soit leur situation financière.
Voici tout ce que vous devez savoir sur Nitisinone et comment y accéder.
Qu'est-ce que le HT-1 ?
La tyrosinémie héréditaire de type 1 (HT-1) est une maladie génétique rare qui affecte le métabolisme d'un acide aminé appelé tyrosine. La tyrosine est présente dans la plupart des protéines.
Les patients atteints de HT-1 présentent une accumulation de déchets de tyrosine dans l'organisme. À terme, cela peut endommager le foie et les reins et augmenter le risque de cancer du foie. Certains patients peuvent également présenter un ramollissement et un affaiblissement des os, ainsi que des problèmes au niveau du système nerveux [1, 2].
Quelle est la rareté du HT-1 ?
L'HT-1 touche environ 1 personne sur 100 000 dans le monde [3]. Il est intéressant de noter que dans certaines régions du Canada, l'incidence est considérablement plus élevée, puisqu'elle touche 1 nouveau-né sur 1 846 [4]. On suppose que cela est dû à une mutation fondatrice dans la population canadienne-française. Une incidence plus élevée de HT-1 est également observée en Turquie et en Inde [4].
Des recherches menées auprès de différentes populations au Royaume-Uni ont suggéré que les personnes d'origine pakistanaise pourraient être plus souvent touchées par l'HT-1 que celles d'origine européenne (3,7 cas d'HT-1 par million contre 0,04 cas par million, respectivement) [5].
Symptômes de la tyrosinémie
Les symptômes de l'HT-1 peuvent apparaître à différents moments de la vie. Certains patients présentent des symptômes dès leur première année, tandis que pour d'autres, ils peuvent apparaître des années plus tard.
Les symptômes diffèrent d'un patient à l'autre et peuvent inclure le rachitisme, une augmentation anormale de la taille de la rate ou du foie, ou une insuffisance hépatique aiguë [2].
Les premiers symptômes de l'HT-1 peuvent être difficiles à détecter. Cependant, le fait de commencer le traitement le plus tôt possible fait une grande différence dans l'évolution de l'état des patients. C'est pourquoi le dépistage néonatal est important [4].
Comment le site Nitisinone traite-t-il la HT-1 ?
Nitisinone facilite la dégradation de la tyrosine. Cela permet de prévenir la toxicité hépatique et rénale, ainsi que les dommages qu'elle provoque [7].
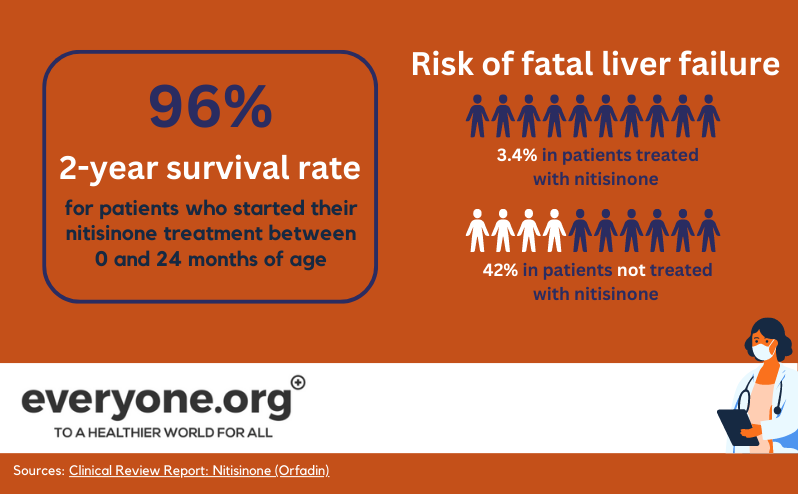
Avant l'introduction du site nitisinone, les patients HT-1 ne vivaient souvent pas jusqu'à l'adolescence. L'insuffisance hépatique ou rénale résultant de la toxicité était fatale pour beaucoup d'entre eux [6]. La transplantation du foie était le seul moyen de corriger le métabolisme de la tyrosine chez les patients HT-1.
Nitisinone Les comprimés d'insuline ont changé la donne. Plus de 90 % des patients répondent très bien au traitement par nitisinone, en association avec un régime pauvre en protéines. Grâce au traitement nitisinone , les greffes de foie ne sont actuellement nécessaires que dans de rares cas [1]. Les patients qui ont commencé le traitement tôt vivent jusqu'à l'âge adulte et ont une vie relativement normale [15].
Nitisinone comme traitement de l'alcalaptonurie (AKU)
L'alcaptonurie (également connue sous le nom de maladie des os noirs, AKU ou maladie des urines noires) est une maladie héréditaire rare qui présente certaines similitudes avec la HT-1. Les patients atteints d'AKU présentent des perturbations dans le métabolisme de l'acide homogentisique oxydase (HGA) - une enzyme qui participe au métabolisme de certains acides aminés tels que la tyrosine et la phénylalanine[11].
Comme les deux partagent une voie métabolique similaire, l'adéquation de nitisinone en tant que traitement de l'alcaptonurie a également été étudiée [12].
D'après les résultats de l'étude, nitisinone semble être un traitement efficace de l'URA chez les patients adultes et une prévention efficace des complications de l'URA chez les enfants [12]. Chez les patients atteints d'UCA également, le traitement par nitisinone doit être associé à une restriction alimentaire des protéines, en particulier de la tyrosine et de la phénylalanine [1].
D'autres études sont nécessaires pour confirmer l'efficacité de nitisinone dans le traitement de l'alcaptonurie, ainsi que pour déterminer les éventuels risques de sécurité pour les jeunes patients [12].
Types de Nitisinone: Orfadin et Nityr
Nitisinone est disponible en tant que médicament générique. Il est vendu sous deux noms de marque : Orfadin et Nityr. Orfadin a été la première version de nitisinone disponible sur le marché. Nityr est bioéquivalent à Orfadin. Cela signifie que les deux médicaments contiennent la même substance active (nitisinone) et agissent de la même manière [8].
Ceci étant dit, il existe quelques différences entre les deux types de traitements nitisinone .
Orfadin vs Nityr: Quelle est la différence ?
Bien qu'Orfadin et Nityr agissent de la même manière et soient prescrits pour traiter la même maladie (HT-1), ils diffèrent l'un de l'autre sur les points suivants :
- Taille. Les comprimés de Nityr sont beaucoup plus petits que les gélules d'Orfadin (environ 20 % de leur taille) [9]. Cela peut faire la différence pour les patients qui ont des difficultés à avaler.
- Conservation. Les gélules d'Orfadin doivent être réfrigérées (ou, si elles sont conservées à température ambiante, elles doivent être utilisées dans les 45 jours ou jetées). Les comprimés Nityr n'ont pas besoin d'être réfrigérés [10]. Ils sont donc plus faciles à emporter.
- Approbation. Seule l'Orfadine est approuvée pour le traitement de l'UCA. Cette autorisation n'est valable que dans l'UE [16]. Cependant, Orfadin et Nityr sont des médicaments bioéquivalents, partageant la même substance active et le même mécanisme d'action. Ce n'est donc qu'une question de temps pour que Nityr obtienne officiellement la même autorisation pour le traitement de l'UCA.
Coût du traitement Nitisinone
Le coût de votre traitement sur nitisinone dépend de plusieurs facteurs, notamment de votre lieu de résidence, du fournisseur de médicaments et de toute couverture d'assurance dont vous pouvez bénéficier.
Le fait que vous utilisiez le médicament pour son usage autorisé (HT-1) ou pour un usage non autorisé (AKU) fait également une différence. Certaines compagnies d'assurance peuvent ne pas couvrir l'utilisation non indiquée d'un médicament.
A titre indicatif, voici les prix approximatifs des comprimés nitisinone :
- Le prix estimé de l'Orfadin pour un mois de traitement (sur la base d'une administration deux fois par jour) peut varier d'environ 5 120 euros à 51 116 euros, en fonction de la puissance requise [12].
- Pour un mois de traitement avec Nityr, les coûts sont estimés entre 5 000 et 25 129 euros, en fonction de la puissance requise [13].
Nitisinone disponible gratuitement en Inde, au Pakistan, au Bangladesh et au Soudan
Le fabricant de Nityr, Cycle Pharma, fournit actuellement des comprimés Nityr gratuitement aux patients AKU et HT-1 en Inde, au Pakistan, au Bangladesh et au Soudan. Ceci est possible grâce à un partenariat établi entre Everyone.org et Cycle Pharma en 2022.
Veuillez noter que certaines conditions peuvent s'appliquer. Si vous êtes basé en Inde, au Pakistan, au Bangladesh ou au Soudan, veuillez soumettre une demande pour Nityr via la page ci-dessous pour savoir si vous avez droit à des comprimés nitisinone gratuits (Nityr).

REMARQUE : Malheureusement, nous ne sommes pas en mesure de livrer au Soudan. Cependant, nous pouvons mettre Nityr à disposition dans l'une de nos pharmacies partenaires au Luxembourg, en Allemagne ou aux Pays-Bas. Contactez-nous pour plus d'informations.
L'avenir des traitements des maladies rares
Bien qu'à l'heure actuelle, seuls 5 % des maladies rares disposent d'un traitement, nous constatons que le paysage médical évolue chaque jour. Nitisinone n'est qu'un exemple parmi d'autres. Nous sommes impatients d'en partager d'autres avec vous.
Références :
- Symptômes et traitement de la tyrosinémie. UPMC Children's Hospital of Pittsburgh, consulté le 2 août 2023.
- Tyrosinémie de type 1 - À propos de la maladie. Genetic and Rare Diseases Information Center, consulté le 2 août 2023.
- Feillet, Francois. Adhésion au traitement dans la tyrosinémie héréditaire de type 1 (HT1) : A Mixed-Method Investigation into the Beliefs, Attitudes and Behaviour of Adolescent Patients, Their Families and Their Health-Care Team. NCBI, 12 septembre 2014.
- Braekeleer, M., et J. Larochelle. Épidémiologie génétique de la tyrosinémie héréditaire au Québec et au Saguenay-Lac-St-Jean. NCBI, consulté le 2 août 2023.
- Une comparaison de la fréquence des maladies et des gènes des erreurs innées du métabolisme entre différents groupes ethniques dans les West Midlands, Royaume-Uni. Journal of Medical Genetics, mai 1998.
-
Das, Martin. Utilité clinique de nitisinone pour le traitement de la tyrosinémie héréditaire de type 1 (HT-1). NCBI, 24 juillet 2017.
-
Nitisinone Oral : Uses, Side Effects, Interactions, Pictures, Warnings & Dosing. WebMD, consulté le 2 août 2023.
-
Nityr. Agence européenne des médicaments, consulté le 2 août 2023.
- Qu'est-ce que Nityr? Nityr, consulté le 2 août 2023.
- ID de référence : 4130114. Accessdata.fda.gov, consulté le 2 août 2023.
- Adéquation du site nitisinone pour la prise en charge de l'alcaptonurie. NCBI, consulté le 2 août 2023.
- Orfadin Prices, Coupons, Copay & Patient Assistance. Drugs.com, consulté le 2 août 2023.
- Nityr Prix, Coupons, Copay & Patient Assistance. Drugs.com, consulté le 2 août 2023.
- Tyrosinémie de type I. Myriad Genetics, consulté le 2 août 2023.
- Succès de DevelopAKUre : approbation de l'Orfadin pour traiter les patients souffrant d'AKU. Recherche et innovation, 26 octobre 2020, consulté le 2 août 2023.